Researchers from the Los Alamos National Laboratory have developed a new computer algorithm that can predict how viruses are going to spread through a human population.
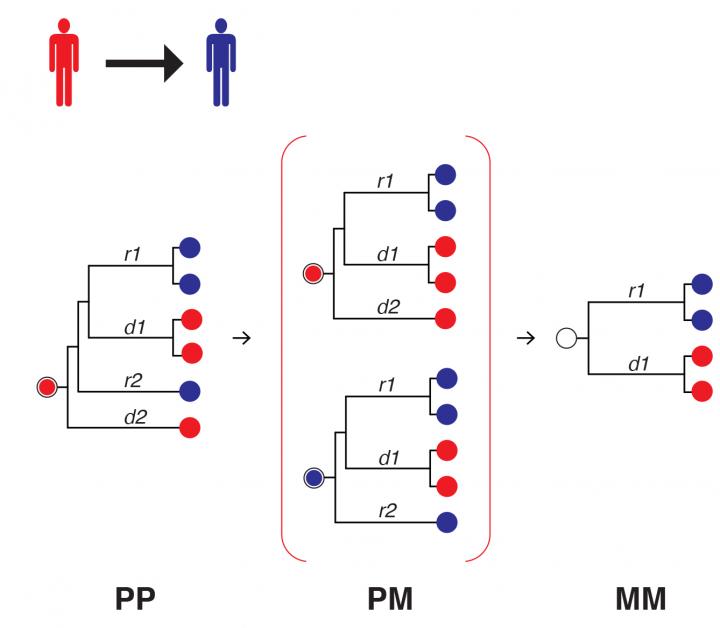 This is the principal decay of paraphyletic signal. (Source: Los Alamos National Laboratory)
This is the principal decay of paraphyletic signal. (Source: Los Alamos National Laboratory)
The simulations from the computer algorithm are consistent with actual DNA that was taken from the global public HIV database at Los Alamos.
"We looked for special genetic patterns that we had seen in the simulations, and we can confirm that these patterns also hold for real data covering the entire epidemic," said Thomas Leitner, a computational biologist at Los Alamos and lead author of the study.
HIV was the focus of this study because it is a rapidly and constantly mutating virus. The changing genetic structures of the virus’ code gives the researchers a clear path that can be followed to determine the origin and time of an infection. The computer simulations have been able to successfully track and predict virus movements through populations of people.
The rapid mutation of the HIV virus is helpful for the epidemiological sleuthing that is behind the development of the computational method and is also one of the reasons it is so difficult to fight the virus with vaccines.
Phylogenetic methods were used to examine evolutionary relationships in the virus’ genetic code to evaluate HIV transmission. These methods found that phylogenetic family tree patterns in the virus correlated to the 955 pairs of people who knew which person was the transmitter and which person was the recipient. The family tree patterns in the virus are the same from the transmitter to the recipient.
"These HIV transmissions had known linkage based on epidemiological information such as partner studies, mother-to-child transmission, pairs identified by contact tracing, and criminal cases," said the authors.
These results led to a collaboration between Colorado and Michigan state health agencies to develop public health computational tools. These tools will help the state agencies track diseases and develop targeted prevention campaigns. The modeling tools could be used to predict patterns of other rapidly evolving infectious diseases.
The paper on this research was published in Nature Microbiology.
